Potencialet bazë termodinamike. Potencialet termodinamike
POTENCIALE TERMODINAMIKE- funksionet e një grupi të caktuar termodinamikë. parametrat, duke ju lejuar të gjeni të gjitha termodinamike. karakteristikat e sistemit në funksion të këtyre parametrave. Të gjitha P.t. janë të ndërlidhura: për secilën prej tyre, me ndihmën e diferencimit në lidhje me parametrat e tij, mund të gjenden të gjitha potencialet e tjera.
Metoda e P.t. u zhvillua nga J. W. Gibbs (J. W. Gibbs) në 1874 dhe është baza e të gjitha termodinamika, duke përfshirë teorinë e sistemeve shumëkomponente, shumëfazore dhe heterogjene, si dhe termodinamike. teori tranzicionet fazore. Ekzistenca e P.t. është pasojë e parimeve të 1-rë dhe të dytë. Statistikore fizika bën të mundur llogaritjen e P. t. bazuar në konceptin e strukturës së materies si sistem i një numër i madh grimcat ndërvepruese.
Energjia e brendshme
U(S, V, N) është një P. t. në rastin kur gjendja e sistemit karakterizohet nga entropia S, vëllimi V dhe numri i grimcave N, e cila është tipike për lëngjet dhe gazet izotropike me një përbërës. U thirrur edhe izokorik-adiabatik. potencial. Diferencial i plotë U barazohet me:
Këtu, variablat e pavarur janë tre të gjera (proporcionale) V) vlerat 5, V, N, dhe ato të varura janë sasitë intensive (të fundme në kufirin termodinamik) që lidhen me të - temperatura. T, presion r dhe potencial kimik Nga kushti që Uështë një diferencial total, rrjedh se variablat e varur T, r, duhet të jenë derivate të pjesshëm të U:
Derivati i dytë U për nga vëllimi jep koeficientin adiabatik. elasticitet:
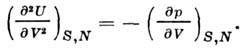
Kapaciteti i nxehtësisë në DC vëllimi është
![]()
Megjithatë, kjo nuk është e vetmja zgjedhje e mundshme e variablave të pavarur që përcaktojnë P. t. Ato mund të zgjidhen nga katër dekomp. mënyrat, kur një termike dhe dy mekanike janë të pavarura. vlerat: S, V, N; S, p, N; T, V, N; T, p, N. Për të zëvendësuar një nga variablat e pavarur me konjugatin e tij në diferencialin total të tipit (1), duhet të kryejmë Transformimi i legjendës, d.m.th., zbres produktin e dy ndryshoreve të konjuguara.
Se. mund të merret entalpia H(S, p, N) (Funksioni termik i Gibbs, përmbajtja e nxehtësisë, potenciali izokorik - izotermik me ndryshore të pavarura S, p, N):
prej nga rrjedh se
Njohuri H ju lejon të gjeni kapacitetin e nxehtësisë në DC. presioni
Energji e lirë
F(T,V,N)(Energjia e Helmholcit, përmbajtja e nxehtësisë, potenciali izobarik-izotermik në variabla T, V, N) mund të merret duke përdorur transformimin Lezhandre të variablave S, V, N te T, V, N:
ku
Derivatet e dyta F sipas V p G jepni kapacitetin e nxehtësisë në DC. vëllimi izotermik. Koeficient presioni
dhe koeficienti izokorik. presioni
Lidhja e fundit bazohet në pavarësinë e derivatit të dytë të përzier të P. t. nga rendi i diferencimit. E njëjta metodë mund të përdoret për të gjetur ndryshimin midis dhe:
dhe raporti ndërmjet adiabatit. dhe izotermike Koeficient kompresim:
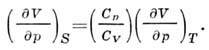
Energjia Gibbs (izobarike - potenciali izotermik në variabla T, p, N) lidhet nga transformimi Lezhandrit në P. t. U, H, F:
ku
proporcionaliteti G numri i grimcave e bën atë shumë të përshtatshëm për aplikime, veçanërisht në teori tranzicionet fazore. Derivatet e dyta G jepni kapacitetin e nxehtësisë në postë. presioni
![]()
dhe izotermike Koeficient ngjeshja
Nga ekuacionet (3), (5), (6), (8) rezulton se P. t. U, H, F, G i lidhur:
to-rye përdoret për të ndërtuar seksion. P. t. sipas ish-perimit. të dhëna termike. dhe kalori. shteti ur-niyah. Kushtet kufitare të nevojshme për këtë jepen nga kalimi në kufi në një gaz ideal dhe Teorema e Nernst-it, i cili konstaton se S=0 brenda kufirit T Oh dhe kështu U=F dhe G - H.
Për sistemet jo të mbyllura, për të cilat N nuk është fikse, është e përshtatshme të zgjidhni P. t. në variabla T, V, e cila nuk ka marrë një emër të veçantë dhe zakonisht shënohet ![]()
Diferenciali i tij total
Të gjitha P. t. janë të lidhura me të ndryshme Shpërndarjet Gibbs. P. t.
lidhur me kanunin e madh. Shpërndarja e Gibbs-it sipas relacionit
ku - integrali statistikor mbi variablat fazore dhe shuma mbi N në rastin e klasikes mekanikë ose funksioni i ndarjes në gjendjet kuantike. P. t. F(T, V, N) lidhet me kanunoren Ansambli Gibbs:
ku është një statistikë. integrale në klasike rasti dhe statistikor sasia në kuantike. P. t. H te shoqeruara me izobarike-izotermike. Ansambli Gibbs, i cili u propozua nga S. A. Boguslavsky (1922). P. t. / 7 shoqërohet me mikrokanonik. Shpërndarja e Gibbs përmes entropisë:
ku W(U, V, N) - statistikë. pesha, to-ry është një faktor normalizimi për mikrokanonik. Shpërndarja e Gibbs. Diferenca totale e entropisë është
që është ekuivalente me ekuacionin (1).
Statistikore integrale ose statistika. shumat në parim mund të llogariten duke u bazuar në f-tionin e Hamiltonit në klasiken. rasti ose operatori Hamiltonian në rastin kuantik për një sistem të një numri të madh grimcash ndërvepruese, e kështu me radhë. llogaritni P. t. me metoda statistikore. mekanika.
Përveç P. t. të listuara, përdoren edhe të tjera, për shembull. Funksionet Massieux - F(T, V, N) IT, funksionet e Planck - ![]() Në rastin e përgjithshëm, kur një sistem me një entropi të caktuar përshkruhet nga një termodinamik parametrat dhe parametrat termodinamikë të lidhur me to. forcat
Në rastin e përgjithshëm, kur një sistem me një entropi të caktuar përshkruhet nga një termodinamik parametrat dhe parametrat termodinamikë të lidhur me to. forcat ![]()

dhe në mënyrë të ngjashme për sistemet me fikse energji.
Për mediat e polarizueshme, P.t. varen nga vektorët elektrikë. dhe magn. induksioni D
dhe AT
. Metoda e P.t. ju lejon të gjeni tensorët e elektricitetit. dhe magn. përshkueshmëria. Në rastin izotropik, dielektriku përshkueshmëria përcaktohet nga ekuacionet

Përdorimi i metodës P.t është veçanërisht efektiv në rastin kur ka lidhje midis parametrave, për shembull. për të studiuar kushtet e termodinamikës. ekuilibri i një sistemi heterogjen i përbërë nga faza të njëpasnjëshme dhe zbërthimi. komponent. Në këtë rast, nëse është e mundur të neglizhohet e jashtme forcat dhe dukuritë sipërfaqësore, krh. energjia e çdo faze është ![]() ku është numri i grimcave të komponentit i në fazë k. Prandaj, për secilën nga fazat
ku është numri i grimcave të komponentit i në fazë k. Prandaj, për secilën nga fazat
(- potenciali kimik i komponentit i në fazë k). P. t. Uështë minimale me kusht që numri i përgjithshëm i grimcave të çdo komponenti, entropia totale dhe vëllimi i çdo faze të mbeten konstante.
Metoda P.t. ju lejon të eksploroni stabilitetin e termodinamikës. ekuilibri i sistemit në lidhje me variacionet e vogla të termodinamikës së tij. parametrave. Ekuilibri karakterizohet nga max. vlera e entropisë ose minimumi i P. t. (energjia e brendshme, entalpia, energjia e lirë, energjia Gibbs), që korrespondon me kushte të pavarura eksperimentale termodinamike. variablave.
Pra, me të pavarur S, V, N për ekuilibër, është e nevojshme që të ketë një int minimal. energji, d.m.th., me ndryshime të vogla në variabla dhe me qëndrueshmëri S, V, N. Prandaj, si kusht i domosdoshëm për ekuilibrin, qëndrueshmëria e presionit dhe e temperaturës së të gjitha fazave dhe barazia e kimikateve. potencialet e fazave bashkëekzistuese. Megjithatë, për termodinamikë qëndrueshmëria nuk mjafton. Nga kushti i minimalitetit të P. t. rrjedh pozitiviteti i variacionit të dytë: > 0. Kjo çon në kushtet e termodinamikës. qëndrueshmëria, p.sh. në një ulje të presionit me rritjen e vëllimit dhe kapacitetit pozitiv të nxehtësisë në DC. vëllimi. Metoda P.t. ju lejon të vendosni për sisteme shumëfazore dhe me shumë komponentë Rregulli i fazës Gibbs, sipas të cilit numri i fazave që bashkëjetojnë në ekuilibër nuk e kalon numrin e përbërësve të pavarur me më shumë se dy. Ky rregull rrjedh nga fakti se numri i parametrave të pavarur nuk mund të kalojë numrin e ekuacioneve për përcaktimin e tyre në ekuilibrin fazor.
Për të ndërtuar një termodinamikë teoritë, të cilat do të merrnin parasysh dukuritë sipërfaqësore, në variacionet e P. t., duhet të merren parasysh termat proporcionalë me ndryshimet në sipërfaqen e fazave kontaktuese. Këto terma janë proporcionale tensioni sipërfaqësor s, që ka kuptim variatet. derivat i ndonjë prej P. t. në lidhje me sipërfaqen.
Metoda P. është gjithashtu e aplikueshme për media të vazhdueshme hapësinore johomogjene. Në këtë rast, P. t. janë funksionale termodinamike. variablave dhe termodinamikë. barazitë marrin formën e ekuacioneve në derivatet funksionale.
Lit.: Vaals I.D. you der, Konstamm F., Kursi i termostatikës, pjesa 1. Termostatik i përgjithshëm, përkth. nga gjermanishtja., M., 1936; Munster A., Termodinamika kimike, përkth. nga gjermanishtja., M., 1971; Gibbs J. B., Termodinamika. Mekanika statistikore, përkth. nga anglishtja, M., 1982; Novikov I.I., Termodinamika, M., 1984. D. N. Zubarev.
Të gjitha llogaritjet në termodinamikë bazohen në përdorimin e funksioneve të gjendjes të quajtura potenciale termodinamike. Çdo grup parametrash të pavarur ka potencialin e vet termodinamik. Ndryshimet në potencialet që ndodhin gjatë çdo procesi përcaktojnë ose punën e bërë nga sistola ose nxehtësinë e marrë nga sistemi.
Kur shqyrtojmë potencialet termodinamike, do të përdorim relacionin (103.22), duke e paraqitur atë në formën
Shenja e barabartë i referohet proceseve të kthyeshme, shenja e pabarazisë - proceseve të pakthyeshme.
Potencialet termodinamike janë funksione të gjendjes. Prandaj, rritja e ndonjërit prej potencialeve është e barabartë me diferencialin total të funksionit me të cilin ai shprehet. Diferenciali total i funksionit të variablave dhe y përcaktohet nga shprehja
![]()
Prandaj, nëse gjatë transformimeve fitojmë një shprehje të formës për rritjen e një vlere të caktuar
mund të argumentohet se kjo sasi është një funksion i parametrave, dhe funksionet janë derivate të pjesshëm të funksionit
Energjia e brendshme. Tashmë jemi njohur me një nga potencialet termodinamike. Kjo është energjia e brendshme e sistemit. Shprehja e parë e ligjit për një proces të kthyeshëm mund të përfaqësohet si
![]() (109.4)
(109.4)
Krahasimi me (109.2) tregon se variablat S dhe V veprojnë si të ashtuquajturat ndryshore natyrore për V potencialin. Nga (109.3) rezulton se
![]()
Nga relacioni del se në rastin - kur trupi nuk shkëmben nxehtësi me mjedisi i jashtëm, puna e bërë prej tij është e barabartë me
![]()
ose në formë integrale:
Kështu, në mungesë të shkëmbimit të nxehtësisë me mjedisin e jashtëm, puna është e barabartë me uljen e energjisë së brendshme të trupit.
në, vëllim konstant
Prandaj, - kapaciteti i nxehtësisë në vëllim konstant është i barabartë me
![]() (109.8)
(109.8)
Energji e lirë. Sipas (109.4), puna e prodhuar nga nxehtësia me të kthyeshme procesi izotermik, mund të përfaqësohet si
Funksioni shtetëror
![]() (109.10)
(109.10)
quhet energjia e lirë e trupit.
Në përputhje me formulat "(109.9) dhe (109.10) në një proces izotermik të kthyeshëm, puna është e barabartë me uljen e energjisë së lirë të trupit:
![]()
Krahasimi me formulën (109.6) tregon se në proceset izotermale energjia e lirë luan të njëjtin rol si energjia e brendshme në proceset adiabatike.
Vini re se formula (109.6) është e vlefshme për proceset e kthyeshme dhe të pakthyeshme. Formula (109.12) është e vlefshme vetëm për proceset e kthyeshme. Me procese të pakthyeshme (shih). Duke e zëvendësuar këtë pabarazi në relacion, është e lehtë të merret se për proceset izotermike të pakthyeshme
Prandaj, humbja e energjisë së lirë përcakton kufirin e sipërm të sasisë së punës që sistemi mund të bëjë në një proces izotermik.
Le të marrim diferencialin e funksionit (109.10). Duke marrë parasysh (109.4) marrim:
Nga një krahasim me (109.2) konkludojmë se ndryshoret natyrore për energjinë e lirë janë T dhe V. Në përputhje me (109.3)
Le të zëvendësojmë: në (109.1) dQ përmes dhe të ndajmë lidhjen që rezulton me ( - kohë). Si rezultat, ne marrim
![]()
Nëse temperatura dhe vëllimi mbeten konstante, atëherë lidhja (109.16) mund të shndërrohet në formë
Nga kjo formulë rezulton se një proces i pakthyeshëm që ndodh në temperaturë dhe vëllim konstant shoqërohet me një ulje të energjisë së lirë të trupit. Kur arrihet ekuilibri, F ndalon së ndryshuari me kalimin e kohës. Në këtë mënyrë; në konstante T dhe V, gjendja e ekuilibrit është gjendja për të cilën energjia e lirë është minimale.
Entalpia. Nëse procesi "ndodh në presion konstant, atëherë sasia e nxehtësisë së marrë nga trupi mund të përfaqësohet si më poshtë:
Funksioni shtetëror
![]()
i quajtur entalpi ose funksioni i nxehtësisë.
Nga (109.18) dhe (109.19) rrjedh se sasia e nxehtësisë së marrë nga trupi gjatë procesit izobatik është e barabartë me
ose në formë integrale
![]()
Prandaj, në rastin kur presioni mbetet konstant, sasia e nxehtësisë së marrë nga trupi është e barabartë me rritjen e entalpisë. Diferencimi i shprehjes (109.19) në lidhje me (109.4) jep
Nga këtu përfundojmë. entalpia është potenciali termodinamik në variabla.Derivatet e tij të pjesshme janë
![]()
Nëse temperatura dhe presioni mbeten konstante, lidhja (109.16) mund të shkruhet si:
Nga kjo formulë rezulton se një proces i pakthyeshëm që ndodh në temperaturë dhe presion konstant shoqërohet me një ulje të potencialit termodinamik Gibbs. Kur arrihet ekuilibri, G pushon së ndryshuari me kalimin e kohës. Kështu, në konstante T dhe gjendja e ekuilibrit është gjendja për të cilën potenciali termodinamik i Gibbs-it është minimal (krh. (109.17)).
Në tabelë. 109.1 tregon vetitë themelore të potencialeve termodinamike.
Tabela 109.1

Ligjëratë me temë: “Potencialet termodinamike”
1. Grupi i potencialeve “E F G H” që kanë dimensionin e energjisë.
2. Varësia e potencialeve termodinamike nga numri i grimcave. Entropia si një potencial termodinamik.
3. Potencialet termodinamike të sistemeve shumëkomponente.
4. Zbatimi praktik i metodës së potencialeve termodinamike (në shembullin e problemit të ekuilibrit kimik).
Një nga metodat kryesore të termodinamikës moderne është metoda e potencialeve termodinamike. Kjo metodë u ngrit kryesisht për shkak të përdorimit të potencialeve në mekanikën klasike, ku ndryshimi i saj shoqërohej me punën e kryer, dhe vetë potenciali është një karakteristikë energjetike e një sistemi termodinamik. Historikisht, potencialet termodinamike të prezantuara fillimisht kishin edhe dimensionin e energjisë, që përcaktoi emrin e tyre.
Grupi i përmendur përfshin sistemet e mëposhtme:
Energjia e brendshme;
Energji e lirë ose potencial Helmholtz;
Potenciali termodinamik i Gibbs;
Entalpia.
Potenciali i energjisë së brendshme u tregua në temën e mëparshme. Ai nënkupton potencialin e sasive të mbetura.
Diferencat e potencialeve termodinamike marrin formën:
Nga relacionet (3.1) mund të shihet se potencialet termodinamike përkatëse karakterizojnë të njëjtin sistem termodinamik me metoda të ndryshme. përshkrimet (metodat e vendosjes së gjendjes së një sistemi termodinamik). Pra, për adiabatik sistem i izoluar, i përshkruar në variabla, është i përshtatshëm për të përdorur energjinë e brendshme si një potencial termodinamik.Pastaj parametrat e sistemit, termodinamikisht të konjuguar me potencialet, përcaktohen nga relacionet:
Nëse një "sistem në një termostat" i dhënë nga variabla përdoret si një metodë përshkrimi, është më e përshtatshme të përdoret energjia e lirë si potencial. Prandaj, për parametrat e sistemit marrim:
Më pas, ne do të zgjedhim modelin "sistem nën piston" si një mënyrë për ta përshkruar atë. Në këto raste, funksionet e gjendjes formojnë një grup (), dhe potenciali Gibbs G përdoret si potencial termodinamik. Pastaj parametrat e sistemit përcaktohen nga shprehjet:
Dhe në rastin e një "sistemi adiabatik mbi një pistoni", dhënë nga funksionet gjendjes, rolin e potencialit termodinamik e luan entalpia H. Pastaj parametrat e sistemit marrin formën:
Meqenëse relacionet (3.1) përcaktojnë diferenciale të plota potencialet termodinamike, mund të barazojmë derivatet e tyre të dytë.
Për shembull, duke pasur parasysh atë
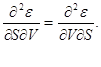
marrim
Në mënyrë të ngjashme, për parametrat e mbetur të sistemit që lidhen me potencialin termodinamik, ne shkruajmë:
Identitete të ngjashme mund të shkruhen edhe për grupe të tjera parametrash të gjendjes termodinamike të sistemit bazuar në potencialin e funksioneve termodinamike përkatëse.
Pra, për një "sistem në një termostat" me një potencial, ne kemi:
Për sistemin "mbi piston" me potencialin Gibbs, barazitë do të jenë të vlefshme:
Dhe, së fundi, për një sistem me një piston adiabatik me potencial H, marrim:
Barazimet e formës (3.6) - (3.9) quhen identitete termodinamike dhe në një numër rastesh rezultojnë të jenë të përshtatshme për llogaritjet praktike.
Përdorimi i potencialeve termodinamike e bën mjaft të lehtë përcaktimin e funksionimit të sistemit dhe efektit termik.
Kështu, relacionet (3.1) nënkuptojnë:
Nga pjesa e parë e barazisë rrjedh dispozita e njohur që puna e një sistemi të izoluar termik () kryhet për shkak të uljes së energjisë së tij të brendshme. Barazia e dytë do të thotë se energjia e lirë është ajo pjesë e energjisë së brendshme, e cila në procesin izotermik shndërrohet plotësisht në punë (përkatësisht, pjesa "e mbetur" e energjisë së brendshme ndonjëherë quhet energji e lidhur).
Sasia e nxehtësisë mund të përfaqësohet si:
Nga barazia e fundit është e qartë pse entalpia quhet edhe përmbajtje nxehtësie. Kur digjet dhe të tjerët reaksionet kimike që ndodh me presion konstant (), sasia e nxehtësisë së çliruar është e barabartë me ndryshimin e entalpisë.
Shprehja (3.11), duke marrë parasysh ligjin e dytë të termodinamikës (2.7), na lejon të përcaktojmë kapacitetin e nxehtësisë:
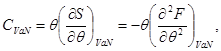

Të gjitha potencialet termodinamike të llojit të energjisë kanë vetinë e aditivitetit. Prandaj, mund të shkruajmë:
Është e lehtë të shihet se potenciali Gibbs përmban vetëm një parametër shtesë, d.m.th. Potenciali specifik i Gibbs nuk varet nga. Pastaj nga (3.4) vijon:
Kjo do të thotë, potenciali kimik është potenciali specifik i Gibbs-it dhe barazia ndodh
Potencialet termodinamike (3.1) janë të ndërlidhura me marrëdhënie të drejtpërdrejta, të cilat bëjnë të mundur kalimin nga një potencial në tjetrin. Për shembull, le të shprehim të gjitha potencialet termodinamike në terma të energjisë së brendshme.
Duke vepruar kështu, ne morëm të gjitha potencialet termodinamike si funksione të (). Për t'i shprehur ato në variabla të tjerë, përdorni procedurën re….
Le të jepet presioni në variabla ():
Shprehjen e fundit le ta shkruajmë si ekuacion të gjendjes, d.m.th. gjeni formularin
Është e lehtë të shihet se nëse gjendja jepet në ndryshore (), atëherë potenciali termodinamik është energjia e brendshme. Në bazë të (3.2), gjejmë
Duke e konsideruar (3.18) si ekuacion për S, gjejmë zgjidhjen e tij:
Duke zëvendësuar (3.19) në (3.17) marrim
Kjo do të thotë, nga variablat () kaluam në variablat ().
Grupi i dytë i potencialeve termodinamike lind nëse, përveç atyre të konsideruara më sipër, potenciali kimik përfshihet si variabla termodinamikë. Potencialet e grupit të dytë kanë gjithashtu dimensionin e energjisë dhe mund të lidhen me potencialet e grupit të parë nga relacionet:
Prandaj, diferencat e mundshme (3.21) kanë formën:
Si dhe për potencialet termodinamike të grupit të parë, për potencialet (3.21) mund të ndërtohen identitete termodinamike, të gjenden shprehje për parametrat e sistemit termodinamik etj.
Le të shqyrtojmë marrëdhëniet karakteristike për "potencialin omega", i cili shpreh energjinë pothuajse të lirë dhe përdoret në praktikë më shpesh midis potencialeve të tjera të grupit (3.22).
Potenciali jepet në variabla () që përshkruajnë sistemin termodinamik me mure imagjinare. Parametrat e sistemit në këtë rast përcaktohen nga marrëdhëniet:
Identitetet termodinamike që vijnë nga potenciali kanë formën:
Mjaft interesante janë vetitë shtuese të potencialeve termodinamike të grupit të dytë. Meqenëse në këtë rast numri i grimcave nuk është ndër parametrat e sistemit, vëllimi përdoret si parametër shtesë. Pastaj për potencialin marrim:
Këtu - potencial specifik për 1. Duke marrë parasysh (3.23), marrim:
Prandaj, (3.26)
Vlefshmëria e (3.26) gjithashtu mund të vërtetohet në bazë të (3.15):
Potenciali mund të përdoret gjithashtu për të kthyer funksionet termodinamike të shkruara në formë në formë. Për këtë, relacioni (3.23) për N:
lejohet në lidhje me:
Jo vetëm karakteristikat energjetike të sistemit, por edhe çdo sasi tjetër e përfshirë në relacionin (3.1) mund të veprojnë si potenciale termodinamike. Si një shembull i rëndësishëm, konsideroni entropinë si një potencial termodinamik. Lidhja fillestare diferenciale për entropinë rrjedh nga shënimi i përgjithësuar i parimeve I dhe II të termodinamikës:
Kështu, entropia është potenciali termodinamik për një sistem i dhënë nga parametrat. Parametrat e tjerë të sistemit duken si:
Duke zgjidhur relacionin e parë (3.28), kalimi nga variablat në variabla është relativisht i mundur.
Vetitë shtuese të entropisë çojnë në marrëdhëniet e njohura:
Le të vazhdojmë me përcaktimin e potencialeve termodinamike në bazë të gjendjeve të dhëna makroskopike të një sistemi termodinamik. Për të thjeshtuar llogaritjet, supozojmë mungesën e fushave të jashtme (). Kjo nuk e zvogëlon përgjithësimin e rezultateve, pasi sistemet shtesë thjesht shfaqen në shprehjet që rezultojnë për .
Si shembull, le të gjejmë shprehje për energjinë e lirë, duke përdorur ekuacionin e gjendjes, ekuacionin kalorik të gjendjes dhe sjelljen e sistemit si ato fillestare. Duke marrë parasysh (3.3) dhe (3.12), gjejmë:
Le të integrojmë ekuacionin e dytë të sistemit (3.30) duke marrë parasysh gjendjen kufitare në:
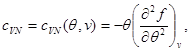
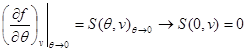

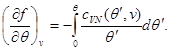
Pastaj sistemi (3.30) merr formën:
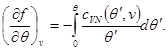
Zgjidhja e sistemit (3.31) bën të mundur gjetjen e energjisë specifike të lirë në formë
Origjina e energjisë specifike të lirë mund të gjendet gjithashtu nga kushtet në:
Pastaj (3.32) merr formën:
dhe shprehja për të gjithë energjinë e lirë të sistemit, deri në një konstante shtesë, merr formën:
Pastaj reagimi i sistemit ndaj përfshirjes së një fushe të jashtme jepet nga një ekuacion shtesë i gjendjes, i cili, në varësi të grupit të ndryshoreve të gjendjes, ka formën:
Pastaj ndryshimi në potencialin termodinamik përkatës të lidhur me përfshirjen e zeros nga zero në përcaktohet nga shprehjet:
Kështu, vendosja e potencialit termodinamik në teorinë makroskopike është e mundur vetëm në bazë të përdorimit ekuacionet e dhëna gjendjet termodinamike, të cilat, nga ana tjetër, fitohen vetë në bazë të vendosjes së potencialeve termodinamike. Thyeje këtë " rrethi vicioz” është e mundur vetëm në bazë të teorisë mikroskopike, në të cilën gjendja e sistemit vendoset në bazë të funksioneve të shpërndarjes, duke marrë parasysh veçoritë statistikore.
Le t'i përgjithësojmë rezultatet e marra në rastin e sistemeve me shumë komponentë. Ky përgjithësim kryhet duke zëvendësuar parametrin me një grup. Le të hedhim një vështrim në shembuj specifikë.
Le të supozojmë se gjendje termodinamike sistemi jepet me parametra, d.m.th. ne konsiderojmë një sistem në një termostat, të përbërë nga disa komponentë, numri i grimcave në të cilat është i barabartë me Energjia e lirë, e cila në këtë përshkrim është potenciali termodinamik, ka formën:
Parametri aditiv në (3.37) nuk është numri i grimcave, por vëllimi i sistemit V. Atëherë dendësia e sistemit shënohet me . Funksioni është një funksion jo shtues i argumenteve jo shtesë. Kjo është mjaft e përshtatshme, pasi kur sistemi ndahet në pjesë, funksioni nuk ndryshon për secilën pjesë.
Pastaj, për parametrat e sistemit termodinamik, mund të shkruajmë:
Duke marrë parasysh që kemi
Për potencialin kimik të një komponenti individual, ne shkruajmë:
Ka mënyra të tjera për të marrë parasysh vetitë shtuese të energjisë së lirë. Le të prezantojmë dendësinë relative të numrit të grimcave të secilit prej përbërësve:

pavarësisht nga vëllimi i sistemit V. Këtu - numri total grimcat në sistem. Pastaj
Shprehja për potencialin kimik në këtë rast merr një formë më komplekse:
Llogaritni derivatet e dhe dhe zëvendësojini ato në shprehjen e fundit:
Shprehja për presion, përkundrazi, do të thjeshtohet:
Marrëdhënie të ngjashme mund të merren edhe për potencialin Gibbs. Pra, nëse vëllimi jepet si një parametër shtesë, atëherë, duke marrë parasysh (3.37) dhe (3.38), shkruajmë:
e njëjta shprehje mund të merret nga (3.yu), e cila në rastin e shumë grimcave merr formën:
Duke zëvendësuar shprehjen (3.39) në (3.45), gjejmë:
që përkon plotësisht me (3.44).
Për të kaluar në regjistrimin tradicional të potencialit Gibbs (përmes variablave të gjendjes ()), është e nevojshme të zgjidhet ekuacioni (3.38):
Në lidhje me vëllimin V dhe zëvendësoni rezultatin në (3.44) ose (3.45):
Nëse numri i përgjithshëm i grimcave në sistemin N jepet si parametër shtesë, atëherë potenciali Gibbs, duke marrë parasysh (3.42), merr formën e mëposhtme:
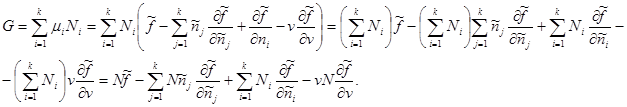
Duke ditur llojin e vlerave specifike: , marrim:
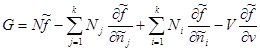
Në shprehjen e fundit, përmbledhja mbi j zëvendësohet me përmbledhjen mbi i. Pastaj termat e dytë dhe të tretë mblidhen deri në zero. Pastaj për potencialin Gibbs më në fund marrim:
E njëjta lidhje mund të merret në një mënyrë tjetër (nga (3.41) dhe (3.43)):
Pastaj për potencialin kimik të secilit prej përbërësve marrim:
Në derivimin e (3.48), transformime të ngjashme me ato të përdorura në derivimin e (3.42) u kryen duke përdorur mure imagjinare. Parametrat e gjendjes së sistemit formojnë një grup ().
Roli i potencialit termodinamik luhet nga potenciali, i cili merr formën:
Siç mund të shihet nga (3.49), i vetmi parametër shtesë në këtë rast është vëllimi i sistemit V.
Le të përcaktojmë disa parametra termodinamikë të një sistemi të tillë. Numri i grimcave në këtë rast përcaktohet nga relacioni:
Për energji të lirë F dhe potencialin Gibbs G mund të shkruhet:
Kështu, marrëdhëniet për potencialet termodinamike dhe parametrat në rastin e sistemeve me shumë komponentë modifikohen vetëm për shkak të nevojës për të marrë parasysh numrin e grimcave (ose potencialet kimike) të secilit komponent. Në të njëjtën kohë, vetë ideja e metodës së potencialeve termodinamike dhe llogaritjeve të bazuara në të mbetet e pandryshuar.
Si shembull i përdorimit të metodës së potencialeve termodinamike, merrni parasysh problemin e ekuilibrit kimik. Le të gjejmë kushtet e ekuilibrit kimik në një përzierje të tre substancave që hyjnë në një reaksion. Për më tepër, ne supozojmë se produktet fillestare të reagimit janë gaze të rrallë (kjo na lejon të injorojmë prodhimin e ndërsjellë ndërmolekular) dhe sistemi ruan temperaturë konstante dhe presioni, (një proces i tillë është më i lehtë për t'u zbatuar në praktikë, prandaj krijohet gjendja e qëndrueshmërisë së presionit dhe temperaturës në instalimet industriale për një reaksion kimik).
Gjendja e ekuilibrit të një sistemi termodinamik, në varësi të mënyrës se si përshkruhet, përcaktohet nga entropia maksimale e sistemit ose energjia minimale e sistemit (për më shumë detaje, shih Termodinamikën Bazarov). Atëherë mund të marrim kushtet e mëposhtme të ekuilibrit për sistemin:
1. Gjendja e ekuilibrit të një sistemi termodinamik të izoluar adiabatikisht, dhënë nga parametrat (), karakterizohet nga një maksimum i entropisë:
Shprehja e dytë në (3.53a) karakterizon qëndrueshmërinë e gjendjes së ekuilibrit.
2. Gjendja e ekuilibrit të një sistemi izokorik-izotermik, e dhënë nga parametrat (), karakterizohet nga një minimum energjie të lirë. Kushti i ekuilibrit në këtë rast merr formën:
3. Ekuilibri i sistemit izobarizotermik, i dhënë nga parametrat (), karakterizohet nga kushtet:
4. Për një sistem në një termostat me një numër të ndryshueshëm grimcash, të përcaktuara nga parametrat (), kushtet e ekuilibrit karakterizohen nga minimumet e mundshme:
Le t'i drejtohemi përdorimit të ekuilibrit kimik në rastin tonë.
Në rastin e përgjithshëm, ekuacioni i një reaksioni kimik shkruhet si:
Këtu janë simbolet substancave kimike, - të ashtuquajturit numra stoikiometrikë. Pra, për reagimin
Meqenëse presioni dhe temperatura janë zgjedhur si parametra të sistemit, të cilët supozohen të jenë konstante. Është e përshtatshme për të konsideruar potencialin Gibbs si një gjendje të potencialit termodinamik G. Atëherë kushti i ekuilibrit për sistemin do të konsistojë në kërkesën e qëndrueshmërisë së potencialit G:
Meqenëse po shqyrtojmë një sistem me tre komponentë, vendosëm Përveç kësaj, duke marrë parasysh (3.54), ne mund të shkruajmë ekuacionin e bilancit për numrin e grimcave ():
Duke prezantuar potencialet kimike për secilin nga komponentët: dhe duke marrë parasysh supozimet e bëra, gjejmë:
Ekuacioni (3.57) u mor për herë të parë nga Gibbs në 1876. dhe është ekuacioni i dëshiruar i ekuilibrit kimik. Është e lehtë të shihet, duke krahasuar (3.57) dhe (3.54), se ekuacioni i ekuilibrit kimik është marrë nga ekuacioni i një reaksioni kimik thjesht duke zëvendësuar simbolet e substancave reaguese me potencialet e tyre kimike. Kjo teknikë mund të përdoret gjithashtu kur shkruhet ekuacioni i ekuilibrit kimik për një reaksion arbitrar.
Në rastin e përgjithshëm, zgjidhja e ekuacionit (3.57), qoftë edhe për tre komponentë, është e ngarkuar mjaftueshëm. Kjo, së pari, për faktin se është shumë e vështirë të merren shprehje të qarta për potencialin kimik edhe për një sistem me një komponent. Së dyti, përqendrimet relative dhe nuk janë sasi të vogla. Kjo do të thotë, është e pamundur të kryhet zgjerimi i serive mbi to. Kjo e ndërlikon më tej problemin e zgjidhjes së ekuacionit të ekuilibrit kimik.
Vështirësitë e theksuara fizikisht shpjegohen me nevojën për të marrë parasysh ristrukturimin predha elektronike atomet që reagojnë. Kjo çon në disa vështirësi në përshkrimin mikroskopik, gjë që ndikon edhe në qasjen makroskopike.
Meqenëse ne ramë dakord të kufizohemi në studimin e rrallimit të gazit, ne mund të përdorim modelin gaz ideal. Supozojmë se të gjithë përbërësit që reagojnë janë gazra idealë që mbushin vëllimin e përgjithshëm dhe krijojnë presion fq. Në këtë rast, çdo ndërveprim (përveç reaksioneve kimike) midis përbërësve të përzierjes së gazit mund të neglizhohet. Kjo na lejon të supozojmë se potenciali kimik i-komponenti i th varet vetëm nga parametrat e të njëjtit komponent.
Këtu - presion i pjesshëm i-komponenti, dhe:
Duke marrë parasysh (3.58), kushti i ekuilibrit për sistemin me tre komponentë (3.57) merr formën:
Për analizë të mëtejshme, ne përdorim ekuacionin e gjendjes së një gazi ideal, të cilin e shkruajmë në formën:
Këtu, si më parë, shënojmë temperaturën termodinamike. Pastaj procesverbali i njohur nga shkolla merr formën: , i cili shkruhet në (3.60).
Pastaj për secilin përbërës të përzierjes marrim:
Le të përcaktojmë formën e shprehjes për potencialin kimik të një gazi ideal. Siç vijon nga (2.22), potenciali kimik ka formën:
Duke marrë parasysh ekuacionin (3.60), i cili mund të shkruhet në formë, problemi i përcaktimit të potencialit kimik reduktohet në përcaktimin e entropisë specifike dhe energjisë specifike të brendshme.
Sistemi i ekuacioneve për entropinë specifike rrjedh nga identitetet termodinamike (3.8) dhe shprehja e kapacitetit të nxehtësisë (3.12):
Duke marrë parasysh ekuacionin e gjendjes (3.60) dhe duke kaluar në karakteristikat specifike, kemi:
Zgjidhja (3.63) ka formën:
Sistemi i ekuacioneve për energjinë e brendshme specifike të një gazi ideal rrjedh nga (2.23):
Zgjidhja për këtë sistem mund të shkruhet si:
Duke zëvendësuar (3.64) - (3.65) në (3.66) dhe duke marrë parasysh ekuacionin e gjendjes për një gaz ideal, marrim:
Për një përzierje gazet ideale shprehja (3.66) merr formën:
Duke zëvendësuar (3.67) në (3.59), marrim:
Duke kryer transformime, ne shkruajmë:

Duke kryer fuqizimin në shprehjen e fundit, kemi:
Relacioni (3.68) quhet ligji i veprimit masiv. Vlera është vetëm një funksion i temperaturës dhe quhet përbërës i një reaksioni kimik.
Kështu, ekuilibri kimik dhe drejtimi i një reaksioni kimik përcaktohet nga madhësia e presionit dhe temperaturës.
Potencialet termodinamike, Schuka, f.36
Potencialet termodinamike, Schuka, f.36
Për sistemet e izoluara, kjo lidhje është ekuivalente me formulimin klasik që entropia nuk mund të ulet kurrë. Ky përfundim u bë nga nobelisti I. R. Prigozhy, duke analizuar sistemet e hapura. Ai gjithashtu avancoi parimin se disekuilibri mund të shërbejë si burim rregulli.
Fillimi i tretë termodinamika përshkruan gjendjen e një sistemi afër zeros absolute. Në përputhje me ligjin e tretë të termodinamikës, ai vendos pikën e referencës së entropisë dhe e rregullon atë për çdo sistem. Në T 0 zhduket koeficienti i zgjerimit termik, kapaciteti termik i çdo procesi. Kjo na lejon të konkludojmë se kur zero absolute temperatura, çdo ndryshim në gjendje ndodh pa ndryshim në entropi. Kjo deklaratë quhet teorema e nobelistit V. G. Nernst, ose ligji i tretë i termodinamikës.
Ligji i tretë i termodinamikës thotë :
zero absolute është thelbësisht e paarritshme sepse në T = 0 dhe S = 0.
Nëse do të kishte një trup me temperaturë të barabartë me zero, atëherë do të ishte e mundur të ndërtohej një makinë e lëvizjes së përhershme e llojit të dytë, e cila bie ndesh me ligjin e dytë të termodinamikës.
Modifikimi i ligjit të tretë të termodinamikës për llogaritjen e ekuilibrit kimik në sistem formuluar nga nobelisti M. Planck në këtë mënyrë.
Postulati i Plankut : në temperaturën zero absolute, entropia merr vlerën S 0 , pavarësisht nga presioni, gjendja e grumbullimit dhe karakteristikat e tjera të substancës. Kjo vlerë mund të vendoset në zero, oseS 0 = 0.
Sipas teorisë statistikore, vlera e entropisë shprehet si S = ln, ku është konstanta e Boltzmanit, – pesha statistikore, ose probabiliteti termodinamik i makrostateve. Quhet edhe -potencial. Nën peshën statistikore nënkuptojmë numrin e mikrogjendjeve, me ndihmën e të cilave realizohet një makrostate e dhënë. Entropia e një kristali ideal në T = 0 K, subjekt i = 1, ose në rastin kur makrostati mund të realizohet nga një mikrogjendje e vetme, është e barabartë me zero. Në të gjitha rastet e tjera, vlera e entropisë në zero absolute duhet të jetë më e madhe se zero.
3.3. Potencialet termodinamike
Potencialet termodinamike janë funksione të grupeve të caktuara të parametrave termodinamikë, duke ju lejuar të gjeni të gjitha karakteristikat termodinamike të sistemit në funksion të këtyre parametrave të njëjtë..
Potencialet termodinamike përcaktojnë plotësisht gjendjen termodinamike të sistemit, dhe çdo parametër i sistemit mund të llogaritet me diferencim dhe integrim.
Potencialet kryesore termodinamike përfshijnë funksionet e mëposhtme .
1. Energjia e brendshme U, i cili është një funksion i variablave të pavarur:
entropia S,
vëllimi V,
numri i grimcave N,
koordinatat e përgjithësuara x i
ose U = U(S, V, N, x i).
2. Energjia e lirë e Helmholcit F është funksion i temperaturës T, vëllimi V, numri i grimcave N, koordinatë e përgjithësuar x i kështu që F = F(T, V, N, x t).
3. Potenciali termodinamik i Gibbsit G = G(T, fq, N, x i).
4. Entalpia H =H(S, P, N, x i).
5. Potenciali termodinamik , për të cilin variablat e pavarur janë temperatura T, vëllimi V, potencial kimik x, = (T, V, N, x i).
Ekzistojnë marrëdhënie klasike midis potencialeve termodinamike:
U = F + TS = H – PV,
F = U – TS = H – TS – PV,
H = U + PV = F + TS + PV,
G = U – TS + PV = F + PV = H – TS,
= U – TS – V = F – N = H – TS – N, (3.12)
U = G + TS – PV = + TS + N,
F = G – PV = + N,
H = G + TS = + TS + N,
G = + PV + N,
= G – PV – N.
Ekzistenca e potencialeve termodinamike janë pasojë e ligjit të parë dhe të dytë të termodinamikës dhe tregojnë se energjia e brendshme e sistemit U varet vetëm nga gjendja e sistemit. Energjia e brendshme e sistemit varet nga grupi i plotë i parametrave makroskopikë, por nuk varet nga mënyra se si arrihet kjo gjendje. Energjinë e brendshme e shkruajmë në formë diferenciale
dU = TdS– PdV– X i dx i + dN,
T = ( U/ S) V, N, x= konst,
P = –( U/ V) S, N, x= konst,
= ( U/ N) S, N, x= konst .
Në mënyrë të ngjashme, dikush mund të shkruajë
dF = – SdT–PdV – X t dx t + dN,
dH= TdS+VdP– X t dx t + dN,
dG= – SdT+VdP – X i dx i + dN,
d = – SdT–PdV – X t dx t – Ndn,
S = – ( F/ T) V ; P = –( F/ V) T ; T = ( U/ S) V ; V = ( U/ P) T ;
S = – ( G/ T) P ; V = ( G/ P) S ; T = ( H/ S;); P = – ( U/ V) S
S = – ( F/ T); N = – ( F/); = ( F/ N); X = – ( U/ x).
Këto ekuacione vlejnë për proceset e ekuilibrit. Le t'i kushtojmë vëmendje potencialit termodinamik izobarizotermik G, thirrur Energjia e lirë e Gibbs,
G = U – TS + PV = H –TS, (3.13)
dhe potenciali izokorik-izotermik
F = U – TS, (3.14)
e cila quhet energjia e lirë e Helmholcit.
Në reaksionet kimike që ndodhin në presion dhe temperaturë konstante,
G = U – TS + PV = N, (3.15)
ku është potenciali kimik.
Nën potencialin kimik të disa komponentëve të sistemit i do të kuptojmë derivatin e pjesshëm të ndonjë prej potencialeve termodinamike në lidhje me sasinë e këtij komponenti në vlera konstante të variablave të tjerë termodinamikë.
Potenciali kimik mund të përkufizohet gjithashtu si një sasi që përcakton ndryshimin në energjinë e sistemit kur shtohet një grimcë e një substance, për shembull,
i = ( U/ N) S , V= kosto , ose G = i N i .
Nga ekuacioni i fundit rezulton se = G/ N i , pra është energjia Gibbs për grimcë. Potenciali kimik matet në J/mol.
Potenciali omega shprehet përmes një funksioni të madh ndarjeje Z si
= – T ln Z, (3.16)
Ku [përmbledhja mbaroi N dhe k(N)]:
Z= exp[( N – E k (N))/T].





