Протонные кислоты примеры. Теория Льюиса
где: K a – константа кислотности; K p – константа равновесия.
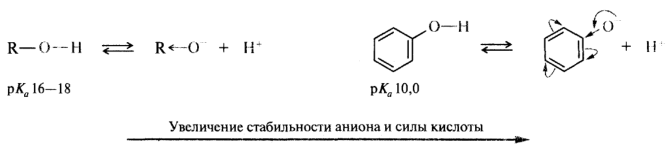
Кислота там сильнее, чем больше константа кислотности. Часто пользуются значениями рК а. Чем меньше величина рК а, тем сильнее кислота.
рК а = - lgК а
Например, рК а фенола = 10, рК а этанола = 16. Это означает, что фенол на шесть порядков (в миллион раз) более сильная кислота, чем этиловый спирт.
Основность может быть выражена через рК b .
рК b = 14 - рК a
Важно помнить, что рК а воды = 15,7. Все вещества, которые имеют рК а больше, чем вода, не способны проявлять кислые свойства в водных растворах. Вода, как более сильная кислота, подавляет диссоциацию более слабых кислот. Так как у большинства органических соединений кислотные свойства выражены во много раз слабее, чем у воды, разработан полярографический подход к оценке их кислотности (И.П. Белецкая и др.). Он позволяет оценивать кислотность до рК а = 50, хотя для очень слабых кислот значения рК а можно оценить только очень приблизительные.
Чрезвычайно важна качественная оценка кислотности как в рядах близких по строению веществ, так и для соединений различных классов. Способность кислоты отдавать протон связана со стабильностью образующегося аниона. Чем стабильнее образующийся анион, тем меньше его стремление захватить протон обратно и превратиться в нейтральную молекулу. При оценке относительной стабильности аниона надо учитывать несколько факторов.
Природа атома, отдающего протон. Атом тем легче теряет протон, чем выше его электроотрицательность и поляризуемость. Поэтому в ряду кислот способность к диссоциации уменьшается следующим образом:
S- H > O- H > - N- H > C- H
Этот ряд прекрасно соответствует свойствам атомов, известным из периодической таблицы.
Влияние окружения. Если сравниваются близкие по строению вещества, оценка проводится сравнением электронной плотности на атоме, отдавшем протон. Все структурные факторы, способствующие уменьшению заряду, стабилизирует анион, а увеличению заряда – дестабилизируют. Таким образом, все акцепторы увеличивают кислотность, все доноры – уменьшают.
Это происходит независимо от того, за счет какого эффекта передачи электронов (индуктивного или мезомерного) происходит перераспределение электронной плотности.
Сольватационный эффект. Сольватация (взаимодействие с молекулами растворителя) повышает стабильность аниона за счет перераспределения избытка электронной плотности между анионом и молекулами растворителя. В общем случае закономерность следующая:
· чем полярнее растворитель, тем сильнее сольватация;
· чем меньше ион, тем лучше он сольватируется.
Основность по Брёнстеду – способность вещества предоставить свою пару электронов для взаимодействия с протоном. Как правило, это вещества, содержащие в молекуле атомы азота, кислорода и серы.
Чем слабее основный центр удерживает пару электронов, тем выше основность. В ряду
R 3 - N > R 2 O > R 2 S
основность уменьшается. Эту последовательность легко запомнить, используя мнемоническое правило “NOS”.
Среди оснований Брёнстеда существует зависимость: анионы более сильные основания, чем соответствующие нейтральные молекулы. Например, гидроксид-анион (– ОН) более сильное основание, чем вода (Н 2 О). При взаимодействии основания с протоном могут образовываться ониевые катионы:
· R 3 О + - оксониевый катион;
· NR 4 + - аммониевый катион;
· R 3 S + - сульфониевый катион.
Качественная оценка основности у близких по строению веществ проводится с использованием той же логики, что и оценка кислотности, но с обратным знаком.
Поэтому все акцепторные заместители основностьи уменьшают, все донорные – увеличивают.
Кислоты и основания по Льюису
Основания по Льюису – доноры электронной пары, как и основания по Брёнстеду.
Определение Льюиса для кислот заметно отличается от привычного (по Брёнстеду). Кислотой по Льюису считается любая молекула или ион, имеющая свободную орбиталь, которая может быть в результате взаимодействия заполнена электронной парой. Если по Брёнстеду кислота – донор протона, то по Льюису сам протон (Н +) – кислота, поскольку его орбиталь пуста. Кислот Льюиса очень много: Na + , Mg 2+ , SnCl 4 , SbCl 5 , AlCl 3 , BF 3 , FeBr 3 и т.д. Теория Льюиса позволяет описать многие реакции как кислотно-основные взаимодействия. Например:
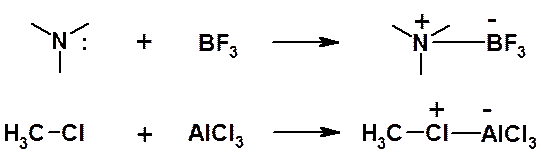
Часто в реакциях с кислотами Льюиса в качестве оснований участвуют органические соединения, являющиеся донорами пары p-электронов:
В органической химии принято следующее:
· если используется термин «кислота» - подразумевается кислота по Брёнстеду;
· если используют термин «кислота» в льюисовском понимании – говорят «кислота Льюиса».
Лекция № 5
Углеводороды
Алканы
· Гомологический ряд, номенклатура, изомерия, алкильные радикалы. Электронное строение молекул алканов, sp 3 -гибридизация, s-связь. Длины C-C и C-H связей, валентные углы, энергии связей. Пространственная изомерия органических веществ. Способы изображения пространственного строения молекул с sp 3 -гибридизованными атомами углерода. Спектральные характеристики алканов. Физические свойства алканов и закономерности их изменения в гомологическом ряду.
Алканы (насыщенные ациклические соединения, парафины)
Алканы - углеводороды с открытой цепью атомов, отвечающие формуле С n H 2 n+2 , где атомы углерода связаны между собой только σ-связями.
Термин «насыщенный» означает, что каждый углерод в молекуле такого вещества связан с максимально возможным числом атомов (с четырьмя атомами).
Строение метана подробно изложено в лекции № 2.
Изомерия, номенклатура
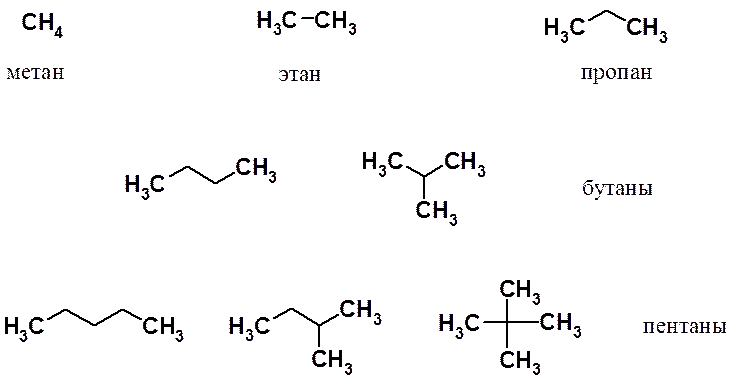
Три первых члена гомологического ряда (метан, этан и пропан) существуют в виде одного структурного изомера. Начиная с бутана число изомеров стремительно растет: у пентана три изомера, а у декана (С 10 Н 22) их уже 75.
Дж. Льюисом была предложена более общая теория кислот и оснований.
Основания Льюиса – это доноры пары электронов (спирты, алкоголят-анионы, простые эфиры, амины и т.д.)
Кислоты Льюиса – это акцепторы пары электронов, т.е. соединения, имеющие вакантную орбиталь (ион водорода и катионы металлов: H + , Ag + , Na + , Fe 2+ ; галогениды элементов второго и третьего периодов BF 3 , AlCl 3 , FeCl 3 , ZnCl 2 ; галогены; соединения олова и серы: SnCl 4 , SO 3).
Таким образом, основания Бренстеда и Льюиса – это одни и те же частицы. Однако основность по Бренстеду есть способность присоединять только протон, в то время как основность по Льюису – понятие более широкое и означает способность к взаимодействию с любой частицей, имеющей низколежащую свободную орбиталь.
Кислотно-основное взаимодействие по Льюису есть доноро-акцепторное взаимодействие и любую гетеролитическую реакцию можно представить как взаимодействие кислоты и основания Льюиса:
Единой шкалы для сравнения силы кислот и оснований Льюиса не существует, так как их относительная сила будет зависеть от того, какое вещество взято за стандарт (для кислот и оснований Бренстеда таким стандартом является вода). Для оценки легкости протекания кислотно-основного взаимодействия по Льюису Р. Пирсоном была предложена качественная теория “жестких” и “мягких” кислот и оснований.
Жесткие основания обладают высокой электроотрицательностью и низкой поляризуемостью. Они трудно окисляются. Их высшие занятые молекулярные орбитали (ВЗМО) имеют низкую энергию.
Мягкие основания имеют низкую электроотрицательность и высокую поляризуемость. Они легко окисляются. Их высшие занятые молекулярные орбитали (ВЗМО) имеют высокую энергию.
Жесткие кислоты имеют высокую электроотрицательность и низкую поляризуемость. Они трудно восстанавливаются. Их низшие свободные молекулярные орбитали (НСМО) имеют низкую энергию.
Мягкие кислоты обладают низкой электроотрицательностью и высокой поляризуемостью. Они легко восстанавливаются. Их низшие свободные молекулярные орбитали (НСМО) имеют высокую энергию.
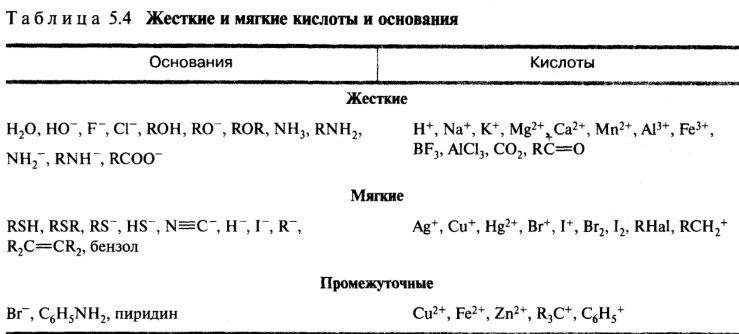
Самая жесткая кислота - Н + , самая мягкая – СН 3 Hg + . Наиболее жесткие основания – F - и OH - , наиболее мягкие – I - и Н - .
Таблица 5. Жесткие и мягкие кислоты и основания.
|
Промежуточные | ||
|
H + , Na + , K + , Mg 2+ , Ca 2+ , Al 3+ , Fe 3+ , BF 3 , AlCl 3 , RC + =O |
Cu 2+ , Fe 2+ , Zn 2+ , R 3 C + |
Ag + , Hg 2+ , I 2 |
|
Основания |
||
|
H 2 O, OH - , F - , ROH, RO - , R 2 O, NH 3 , RNH 2 |
ArNH 2 , Br - , C 5 H 5 N |
R 2 S, RSH, RS - , I - , H - , C 2 H 4 , C 6 H 6 |
Принцип жестких и мягких кислот и оснований Пирсона (принцип ЖМКО):
Жесткие кислоты преимущественно взаимодействуют с жесткими основаниями, а мягкие кислоты – с мягкими основаниями.
Это выражается в больших скоростях реакций и в образовании более устойчивых соединений, так как взаимодействие между близкими по энергии орбиталями эффективнее, чем взаимодействие между орбиталями, значительно различающимися по энергии.
Принцип ЖМКО используют для определения преимущественного направления конкурирующих процессов (реакции элиминирования и нуклеофильного замещения, реакции с участием амбидентных нуклеофилов); для направленного создания детоксикантов и лекарственных препаратов.
Классическую электронную теорию химической связи предложил в 1916 г. американский химик Гилберт Ньютон Льюис (1875—1946). Как уже было известно, завершённые электронные оболочки атомов благородных газов отличаются особенной устойчивостью. Льюис предположил, что при образовании химической связи возникают пары электронов, которые принадлежат сразу двум атомам. Тем самым атомы заполняют до конца свой внешний электронный уровень и приобретают электронную конфигурацию благородного газа. Льюис назвал это правилам октета, так как атомы всех благородных газов (кроме гелия) имеют на внешнем уровне восемь электронов. С помощью правила октета удалось объяснить электронное строение огромного числа соединений. А связь, образованная за счёт общей пары электронов, получила название ковалентной.
Для обозначения электронов Льюис использовал точки: Н:Н. Но часто общую пару электронов изображают просто чёрточкой, которая и символизирует химическую связь: Н—Н. В молекуле Н 2 каждому атому принадлежат два электрона (конфигурация атома гелия), которые предоставлены в общее пользование двумя атомами водорода (это обменный механизм образования ковалентной связи).
Подобным образом устроена и молекула F 2 . У атома фтора на внешнем уровне семь электронов — чтобы достичь электронной конфигурации неона не хватает одного. Поэтому каждый атом F отдаёт по одному электрону в общее пользование:
Теперь оба атома имеют на внешнем уровне по восемь электронов (октет), из которых два общих, а шесть (три пары) сохраняются в индивидуальном пользовании.
Аналогично образуются и кратные связи. Так, тройная связь в молекуле N 2 (NºN) возникает в результате создания трёх общих электронных пар.
В двухатомных молекулах простых веществ (Н 2 , F 2 , N 2 и др.) общие пары электронов в равной степени принадлежат обоим атомам. Такая связь называется непопярной. При образовании ковалентной связи в молекулах сложных веществ общая электронная пара оказывается смещённой в сторону одного из атомов. Молекула при этом поляризуется: одна её часть несёт частичный положительный заряд (8+), а другая — отрицательный (8-). Примером может служить молекула HF, в которой атом Н достигает электронной конфигурации гелия, а атом F — неона:
Общая электронная пара в этом соединении смещена в сторону атома фтора. Это пример полярной связи.
Бывает, что один из атомов (донор электронов) предоставляет в общее пользование два электрона, а другой (акцептор) — ни одного. Такой механизм образования ковалентной связи называют донорно-акцепторным (иногда эту связь обозначают стрелкой, направленой к акцептору).
Пример
— ион аммония. Атом азота имеет три неспаренных электрона, которые
участвуют в трёх ковалентных связях с тремя атомами водорода. Кроме
того, у атома азота есть ещё одна пара электронов. При присоединении
иона Н + к аммиаку NH 3 эта
пара поступает в совместное пользование атомов азота и водорода. В
данном случае атом азота выступает в качестве донора, а Н + — акцептора электронной пары:
В молекуле С=О атомы связаны тройной связью: две из этих связей образованы по обменному механизму, а третья — по донорно-акцепторному.
Теория Льюиса позволяет не только описать распределение электронов в молекулах, но и рассчитать так называемые эффективные заряды на атомах. Для этого электронный «колхоз» временно «разгоняют», электроны, участвующие в химической связи, делят поровну между атомами, подсчитывают общее число электронов каждого атома и сравнивают его с числом валентных электронов до образования связи. В молекуле СО на долю углерода приходится пять электронов (два своих и три из шести общих), тогда как в свободном атоме углерода — четыре электрона. Лишний электрон означает, что эффективный заряд на атоме углерода в молекуле СО равен -1. Молекула в целом электронейтральна, поэтому заряд на атоме кислорода равен +1.
В общем виде кислотно-основное взаимодействие описывается уравнением
В принципе большинство органических соединений можно рассматривать как потенциальные кислоты, поскольку в них содержатся атомы водорода, связанные с разными элементами (О, S, N, С). Элемент и связанный с ним атом водорода называют кислотным центром . Органические кислоты соответственно классифицируют по кислотному центру как ОН-, SH-, NH- и СН-кислоты. Кислотами могут быть не только нейтральные молекулы, но и положительно заряженные ионы, а также диполярные ионы. Органические основания для образования ковалентной связи с протоном кислоты должны либо иметь неподеленную пару электронов у гетероатома (нейтральные молекулы), либо быть анионами. В целом основания, имеющие в молекулах гетероатом, называются n -основаниями . Существует еще одна группа оснований - π -основания , в которых центром основности являются электроны локализованной π-связи или π-электронного облака сопряженной системы. π -Основания образуют с протоном не ковалентные связи, а короткоживущие π -комплексы.
Кислотность и основность веществ по Брёнстеду-Лоури характеризуется количественно. Применяя закон действующих масс, можно выразить кислотные свойства кислоты А-Н через константу равновесия K p , представленной выше реакции обратимого кислотно-основного взаимодействия:
![]()
Очевидно, что константа равновесия реакции ионизации кислоты имеет постоянное значение только для данной системы и по отношению к каждому основанию существует своя шкала констант кислотности. Наиболее важным случаем является ионизация кислот в водном растворе (вода играет роль основания):
Поскольку вода присутствует в большом избытке, то ее концентрация остается практически постоянной, равной 55,5 моль/л. Это значение включают в константу равновесия и получают характеристику, называемую константой кислотности К а :
Чем больше К а , тем сильнее кислота . Однако даже такая сравнительно сильная по меркам органических соединений кислота, как уксусная, имеет К а = 1,75 10 -5 . Для большинства органических соединений К а имеют еще меньшие значения. Поэтому для оценки силы органических кислот значительно удобнее пользоваться значениями р К а представляющими собой отрицательный логарифм констант кислотности: р К а = -lg К а . При этом чем меньше р К а , тем сильнее кислота . Кислоты, у которых рК а > 7, не изменяют цвет нейтральной индикаторной бумаги; кислоты с рК а >10 не имеют кислого вкуса.
Основность соединений в водном растворе можно охарактеризовать величиной рК b , которая связана с рК а через ионное произведение воды: р К b = 14 - р К а . Однако в настоящее время для характеристики основности чаще используют величину рК а сопряженной основанию В кислоты ВН + , обозначаемую как р K BH + . Такой подход позволяет применять одну и ту же шкалу для характеристики ионизации как кислот, так и оснований. В этом случае чем больше pK BH + , тем сильнее основание .
Слабые кислоты и основания в биологических системах. Большинство биологически активных органических соединений, в частности лекарственных веществ, являются слабыми кислотами или основаниями. Степень ионизации таких соединений в той или иной среде имеет важное значение для проявления биологического действия. Известно много лекарственных веществ, терапевтическая активность которых определяется долей присутствующих неионизированных молекул, хотя существуют и другие примеры, когда, наоборот, ионизированная часть вещества обусловливает биологический эффект за счет взаимодействия с катионными или анионными центрами рецепторов. Различия в степени ионизации обеспечивают избирательность действия, и это связано с такими факторами, как, например, проникновение через мембраны в плазму крови или клетку, адсорбцией на поверхностях ферментов, возможной ионизацией центров рецептора в зависимости от pH и т. д.

Степень ионизации органических кислот и оснований в растворе определяется значениями двух параметров: pH раствора и рК а кислоты (или рК BH + основания). Если значения рК а (или pK BH +) вещества и pH раствора известны, то степень ионизации может быть рассчитана следующим образом:

Степень ионизации имеет важное значение для процессов проникновения веществ через различные мембраны в организме, например при всасывании (абсорбции) лекарств из желудочно-кишечного тракта. Мембраны эпителия пищеварительного тракта можно рассматривать как липидный бислой, в который встроены белковые молекулы. Гидрофобные участки мембранных белков погружены во внутреннюю полость мембраны, а ионизированные участки обращены к водной фазе внутри и снаружи. Согласно классической теории, мембраны подобного типа препятствуют прохождению ионов, так как, во-первых, ионы вследствие гидратации имеют относительно большой размер и, во-вторых, если заряд иона и заряд белковой поверхности, к которой он приближается, аналогичны по знаку, то происходит отталкивание, а если противоположны, то происходит адсорбция иона на поверхности мембраны. Через природные мембраны проникают только те ионы, для которых существуют специфические транспортные системы или переносчики. Нейтральные липидорастворимые молекулы проникают через мембраны и тем быстрее, чем выше их липофильные свойства. Таким образом в желудочно-кишечном тракте происходит всасывание неионизированных молекул лекарственных веществ.
Препараты кислотной природы будут лучше всасываться из желудка (pH 1-3), а всасывание лекарств-оснований будет происходить только после того, когда они пройдут из желудка в кишечник (содержимое тонкого кишечника имеет pH 7-8). В течение одного часа из желудка крыс всасывается почти 60% ацетилсалициловой кислоты и только 6% анилина от введенной дозы. В кишечнике крыс всасывается уже 56% от введенной дозы анилина. Примечательно, что такое слабое основание, как кофеин (рK В H + 0,8), всасывается за то же время в гораздо большей степени (36%), так как даже в сильнокислой среде желудка кофеин в значительной степени находится в неионизированном состоянии.
Эффективность действия лекарственных веществ обусловливается способностью их проникновения к рецептору. Дли веществ, способных к ионизации, биологическая активность может определяться долей неионизированных молекул или, наоборот, ионизированной частью вещества. Имеются многочисленные примеры и того, и другого вариантов. Так, и фенол и уксусная кислота прекращают рост различных плесневых грибов; их биологическое действие обусловлено неионтированными молекулами, и поэтому наибольшая эффективность уксусной кислоты проявляется при pH ниже 4, а для фенола при любых значениях pH ниже 9, так как в этих диапазонах значений pH и фенол и уксусная кислота находятся в неионизированном состоянии. Также только неионизированный теофиллин, в отличие от своего аниона, стимулирует деятельность сердца черепахи. На примере ряда сульфаниламидных препаратов, наоборот, установлено, что их антибактериальная активность обусловлена анионами. Оптимальное для проявления активности значение рК а сульфаниламидов находится в интервале 6-8. Через мембрану в клетку проникают неионизированные молекулы, но при физиологических значениях pH вновь образуются ионы, пока по обе стороны мембраны не установится равная степень ионизации:
Антибактериальная активность сульфаниламидов пропорциональна степени ионизации, но зависит также и от липофильности молекул.
И еще один пример, когда биологическая активность обусловлена ионизированной формой вещества: антибактериальное (бактериостатическое) действие аминоакридинов проявляется только в катионной форме этих соединений и возрастает при повышении степени их катионной ионизации.Изменение степени ионизации в зависимости от pH среды широко используют для выделения лекарственных веществ из биологических жидкостей (кровь, моча) с целью их последующего анализа, например, при проведении фармакокинетических исследований.
Кислоты и основания Льюиса . Согласно теории Льюиса, кислотно-основные свойства соединений определяются их способностью принимать или отдавать пару электронов с образованием новой связи. Кислоты Льюиса - акцепторы пары электронов. Основания Льюиса доноры пары электронов.
Основания Брёнстеда и основания Льюиса являются донорами пары электронов - либо неподеленной, либо находящейся на р-орбитали, т. е. понятия тождественны в той и другой теории. Кислотность по Льюису имеет новый и более широкий смысл. Кислотой считается любая частица с вакантной орбиталью, которая способна дополнить свою электронную оболочку парой электронов. По Брёнстеду кислота - это донор протона, а по Льюису - сам протон Н + является кислотой, так как имеет вакантную орбиталь.
Кислотами Льюиса являются галогениды элементов второй и третьей групп периодической системы (BF 3 , А1С1 3 , FeCl 3 , FeBr 3 , ZnCl 2 и др.). К кислотам Льюиса также относятся галогениды других элементов, имеющих вакантные орбитали - SnX 4 , SbX 5 , AsX 5 и даже оксид серы (VI) SО 3 . Галогениды бора, алюминия имеют по шесть электронов на внешней оболочке и способны принимать пару электронов с образованием ковалентной связи. Тетрахлорид олова, например, имеет 8 электронов на внешней оболочке, но как элемент, имеющий вакантные орбитали, способен принять еще пару электронов. К кислотам Льюиса относятся также катионы металлов (Na + , Mg 2+ , Ag +), карбокатионы R 3 C + , нитроил-катион NО 2 + и др. В гетеролитических реакциях кислоты Льюиса участвуют как электрофильные реагенты. Ниже приведены некоторые примеры взаимодействия между кислотами и основаниями Льюиса:
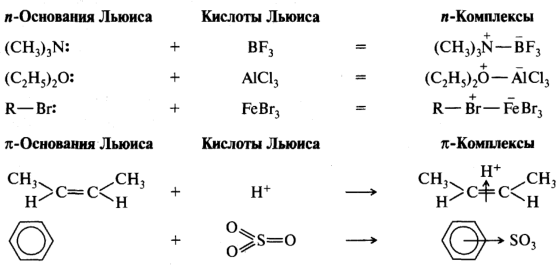
Многие распространенные органические реакции относятся к кислотноосновным взаимодействиям в рамках теории Льюиса. Однако в этой теории гораздо сложнее дать количественную оценку кислотности и основности, и такая оценка может быть лишь относительной. Для этого определяют энергии взаимодействия различных соединений в строго определенных условиях (растворитель, температура) с одним и тем же стандартом, являющимся соответственно кислотой или основанием Льюиса. Поэтому количественных измерений для кислот и оснований Льюиса сделано намного меньше, чем для кислот и оснований Брёнстеда.
Жесткие и мягкие кислоты и основания. Развитие теории Льюиса привело к созданию принципа жестких и мягких кислот и оснований (принцип ЖМКО, принцип Пирсона). Согласно принципу Пирсона, кислоты и основания подразделяются на жесткие и мягкие.
Жесткие кислоты - это кислоты Льюиса, в которых акцепторные атомы малы по размеру, обладают большим положительным зарядом, большой электроотрицательностью и низкой поляризуемостью. Мягкие кислоты Льюиса содержат акцепторные атомы большого размера с малым положительным зарядом, небольшой электроотрицательностью и высокой поляризуемостью.
Низшая свободная молекулярная орбиталь (НСМО), которая участвует в связывании с орбиталью донора пары электронов, у жестких кислот имеет низкую энергию. Самой жесткой кислотой является протон. НСМО мягких кислот имеет высокую энергию. Мягкие кислоты содержат легко поляризуемые вакантные орбитали. Положительный заряд у атома - акцептора пары электронов мал вследствие делокализации или вообще отсутствует (так, мягкой кислотой является молекула иода).
Жесткие основания - это донорные частицы, обладающие высокой электроотрицательностью, низкой поляризуемостью, трудно окисляющиеся. Мягкие основания , напротив, - это донорные частицы с низкой электроотрицательностью, высокой поляризуемостью, довольно легко окисляющиеся. Термин «жесткое основание» подчеркивает, что соединение - донор пары электронов - прочно удерживает свои электроны. У жестких оснований высшая занятая молекулярная орбиталь (ВЗМО), которая участвует в связывании с орбиталью акцептора пары электронов, имеет низкую энергию (расположена близко к ядру атома). Атомами-донорами в жестких основаниях являются азот, кислород, фтор, хлор. Мягкие основания слабо удерживают свои валентные электроны, ВЗМО донора имеет высокую энергию. Донорами пары электронов выступают атомы углерода, серы, фосфора, иода.
Следует отметить, что понятия «жесткие» и «мягкие» кислоты и основания не равноценны понятиям «сильные» и «слабые» кислоты и основания. Это две независимые характеристики кислот и оснований. Принцип ЖМКО используется для качественного описания эффективности протекания кислотно-основного взаимодействия: (!) жесткие кислоты лучше координируются с жесткими основаниями, мягкие кислоты - с мягкими основаниями. Концепция Пирсона основана на том, что взаимодействие между орбиталями с близкими энергиями более эффективно, чем между орбиталями, имеющими разную энергию.
Действие принципа ЖМКО можно проиллюстрировать следующим примером. При взаимодействии галогеноалканов с нуклеофилами (являющимися также и основаниями) могут происходить конкурентные реакции - нуклеофильного замещения или элиминирования. Реакция нуклеофильного замещения осуществляется посредством взаимодействия нуклеофила с атомом углерода, связанным с галогеном. В реакции элиминирования происходит также и отщепление протона от соседнего атома углерода под влиянием основания.
При взаимодействии 1,2-дихлороэтана с жестким основанием (метоксид-ионом) вследствие атаки реагента на жесткую кислоту - протон преимущественно происходит реакция элиминирования. Мягкое основание - тиофеноксид-ион - предпочтительно реагирует с более мягкой кислотой - атомом углерода, в результате чего образуется продукт реакции нуклеофильного замещения:
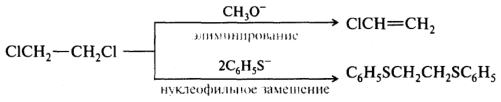
Типы органических кислот. Факторы, определяющие кислотность. Примеры.
Способность органических соединений к ионизации изменяется в широких пределах. В водном растворе экспериментально возможно определить рК а лишь до значений -15 (рК а воды 15,7). Для более слабых кислот определение рК а проводят в других растворителях. Например, в жидком аммиаке можно определять рК а до значения 33. Соотнести кислотность в воде и аммиаке можно, определив рК а для какого-либо соединения параллельно в этих двух средах, что позволяет осуществлять переход от одной шкалы к другой. Подход к оценке кислотности очень слабых СН-кислот - предложили полярографическую шкалу кислотности. Эта шкала охватывает кислоты, у которых кислотность изменяется в пределах 50 порядков. И все же полученные значения рК а для слабых СН-кислот являются весьма приблизительными.
Ввиду необъятно большого числа органических соединений невозможно для каждого из них иметь количественную оценку кислотных свойств. Действительно, константы рК а в различных растворителях установлены для относительно небольшого числа соединений и неизвестны для многих даже важных биологически активных веществ. Поэтому большое значение приобретает качественный подход к оценке кислотных свойств разных кислотных центров, который базируется на оценке стабильности сопряженного основания : (!) сила кислоты определяется стабильностью сопряженного основания (аниона), образующегося из этой кислоты. Чем стабильнее анион, тем сильнее кислота.
Другими словами, кислотность зависит от совокупности ряда факторов, обусловливающих стабильность аниона А - :
электроотрицательности и поляризуемости элемента, отдающего протон;
степени делокализации отрицательного заряда в анионе;
способности аниона к сольватации, т. е. взаимодействию с молекулами растворителя.
Обычно на кислотность большинства веществ в растворе оказывают влияние одновременно несколько факторов, но в каждом конкретном случае один или несколько из них будут преобладающими. Ниже будет рассмотрена роль этих факторов в определении стабильности анионов (вначале без учета влияния среды). В отсутствие эффектов сольватации проявляется истинная (собственная) кислотность данного соединения. Собственная кислотность проявляется в газовой фазе, и в этом случае она определяется исключительно структурой соединения.
Природа атома в кислотном центре. Роль электроотрицательности и поляризуемости элемента в кислотном центре может быть наглядно продемонстрирована на примере кислот Брёнстеда с различными кислотными центрами, но с одинаковыми заместителями, в данном случае этильными радикалами:
По возрастанию кислотности кислоты Брёнстеда:
В таком же порядке возрастает стабильность соответствующих анионов. Чем более электроотрицательным является элемент в кислотном центре, тем он более способен нести отрицательный заряд и соответственно тем стабильнее будет образующийся анион .
Поскольку электроотрицательность атома кислорода (3,5) больше, чем атома азота (3,0) и атома углерода (2,5), то в таком же порядке будет уменьшаться стабильность соответствующих анионов. Сравниваемые элементы находятся в одном периоде, и их поляризуемость практически одинакова. Для элементов третьего и последующего периодов периодической системы основное влияние на стабильность аниона оказывает фактор поляризуемости . В приведенной выше группе кислот Брёнстеда в случае этантиол а атом серы больше по размеру и легче поляризуется, чем элементы второго периода (О, N, С) у других кислот. Отрицательный заряд на атоме серы делокализован в большей степени. Поэтому алкантиолят-ион стабильнее, чем соответствующий алкоксид-ион и т. д. В целом же SH-кислота будет сильнее ОН-, NH- и СН-кислот. Для представленной выборки соединений кислотность в газовой фазе и в растворе будет одинаковой в связи с тем, что сольватация близких по размеру анионов будет нивелирована. Тиолы, как более сильные кислоты, реагируют со щелочами, а также с оксидами, гидроксидами и солями тяжелых металлов. Со щелочными металлами тиолы образуют растворимые в воде соли, с тяжелыми металлами - нерастворимые:
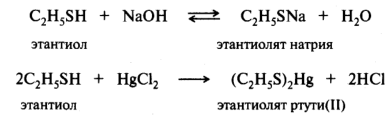
Способность тиолов связывать ионы тяжелых металлов обусловила использование их в качестве противоядия при отравлениях соединениями мышьяка, ртути, хрома, висмута и других металлов, относящихся к тиоловым ядам.
Спирты, как слабые кислоты, практически не реагируют с гидроксидами металлов; значения рК а спиртов близки рК а воды, равной 15,7. При взаимодействии этанола со щелочью равновесие сдвинуто в сторону исходного спирта и содержание этоксида натрия в реакционной смеси будет невелико. Однако спирты способны реагировать со щелочными металлами и сильными основаниями, такими, как гидриды или амиды металлов, литий- и магнийорганические реагенты:
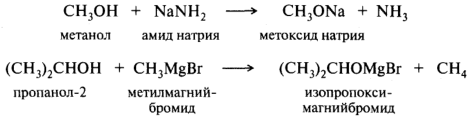
Этиламин и пропан не проявляют заметных кислотных свойств. Тем не менее в других NH- и СН- кислотные свойства выражены гораздо сильнее, что обусловлено электроноакцепторным влиянием заместителей, связанных с кислотным центром.
Стабилизация аниона за счет сопряжения. При сравнительной оценке кислотности соединений, имеющих одинаковый элемент в кислотном центре, основным фактором, определяющим собственную кислотность, становится делокализация отрицательного заряда в анионе. Стабильность аниона значительно повышается, если имеется возможность делокализации отрицательного заряда по системе сопряженных связей. Характерным примером проявления действия этого фактора в группе ОН-кислот является повышение кислотности при переходе от спиртов к фенолам и к карбоновым кислотам.
Увеличение кислотности фенолов по сравнению с алифатическими спиртами объясняется большей стабильностью феноксид-иона, в котором отрицательный заряд делокализуется с участием атомов углерода бензольного кольца:
Повышенная по сравнению с фенолами кислотность карбоновых кислот обусловлена стабилизацией ацилат-ионов, в которых отрицательный заряд за счет р, π-сопряжения распределен поровну между двумя атомами кислорода:
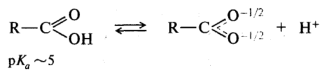
Делокализация отрицательного заряда по системе сопряженных связей, приводящая к стабилизации аниона, приводит к увеличению кислотности и других типов кислот.
Влияние электронных эффектов заместителей, связанных с кислотным центром. Независимо от механизма передачи электронного влияния заместителя (индуктивного или мезомерного) в общем случае выполняется нижеприведенное правило: (!) Электроноакцепторные заместители способствуют делокализации отрицательного заряда, стабилизируют анион и тем самым увеличивают кислотность. Электронодонорные заместители, наоборот, понижают ее. Влияние на кислотность электроноакцепторных атомов галогенов наглядно иллюстрируется значениями рК а моно- и тригалогенозамещенных уксусных кислот. Наиболее сильный эффект оказывает самый электроотрицательный элемент фтор:
Влияние заместителей особенно ярко проявляется в ряду замещенных фенолов. Электроноакцепторная нитрогруппа, например, дополнительно стабилизирует образующийся анион, что приводит к увеличению кислотности n-нитрофенола (рК а 7,1) по сравнению с незамещенным фенолом (рК а 10). Наличие в бензольном кольце трех нитрогрупп приводит к тому, что 2,4,6-тринитрофенол (пикриновая кислота) становится уже очень сильной кислотой (рК а 0,8), сравнимой с минеральными кислотами. Электронодонорные метальная и аминогруппы дестабилизируют феноксид-ионы и уменьшают кислотность n-метилфенола (рК а 10,1) и n-аминофенола (рК а 10,5):
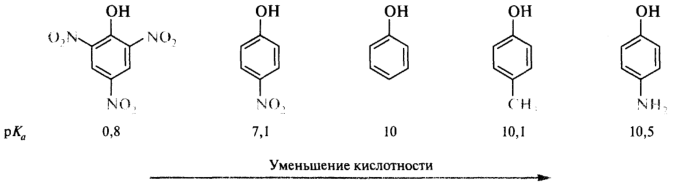
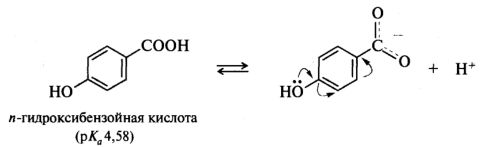
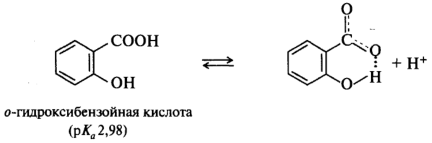
В ароматических кислотах влияние заместителей, находящихся в мета- и пара -положениях бензольного кольца, подчиняется общему правилу: электроноакцепторные - увеличивают кислотность, электронодонорные - уменьшают. Поведение орто-замещенных кислот часто бывает аномальным. Как правило, орто-замещенные бензойные кислоты сильнее соответствующих пара-изомеров, независимо от того, является ли заместитель донором или акцептором. Такое влияние заместителя называют орто-эффектом. Иногда орто -эффект имеет вполне очевидное объяснение. Например, n-гидроксибензойная кислота (рK a 4,58) слабее бензойной (рK a 4,19), как и ожидалось исходя из влияния на кислотность электронодонорной ОН-группы. Однако салициловая (о-гидроксибензойная) кислота гораздо сильнее (рК а 2,98), поскольку в стабилизацию образующегося из этой кислоты салицилат-иона вносит вклад внутримолекулярная водородная связь, что и приводит к увеличению кислотности именно этого изомера:
Эффект сольватации. Влияние сольватации может быть очень значительным. Почти во всех случаях кислотно-основных взаимодействий можно считать, что исходные нейтральные молекулы и образующиеся ионы сольватируются по-разному. Стабильность аниона существенно зависит от его сольватации в растворе. Взаимодействие между растворителем и ионом может быть различным по своей природе - электростатическим, координационным (в том числе и за счет водородных связей), гидрофобным. При сольватации иона происходит перераспределение заряда с участием окружающих его молекул растворителя. Как правило, сольватация ионов в полярных растворителях тем сильнее, чем более полярен растворитель . Поскольку всю совокупность взаимодействий между ионом и окружающей его средой учесть чрезвычайно трудно, то обычно пользуются эмпирическим правилом : (!) чем меньше размер иона и чем больше локализован в нем заряд, тем он лучше сольватируется.
Трудным для интерпретации является вопрос о соотношении между кислотностью соединений в водной среде и газовой фазе. Развитие методов ионного циклотронного резонанса и масс-спектрометрии высокого давления обеспечило возможность достаточно точных определений термодинамических равновесий в газовой фазе. Так, было установлено, что по силе кислотности уксусная кислота и фенол в газовой фазе близки, тогда как в воде рК а этих соединений за счет эффекта гидратации анионов различаются на пять порядков. Полагают, что делокализация отрицательного заряда по бензольному кольцу в фенолят-ионе снижает его способность к образованию водородных связей с водой. Собственная кислотность алифатических спиртов в газовой фазе возрастает с увеличением длины и разветвленности алкильного радикала.
Дж. Льюисом (1923) была предложена более общая теория кислот и оснований, опирающаяся на строение внешних электронных оболочек атомов. По теории Льюиса кислотные и основные свойства соединений определяются их способностью принимать или отдавать пару электронов с образованием связи.
Кислотами Льюиса могут быть атом, молекула или катион, обладающие вакантной орбиталью и способные принимать пару электронов с образованием ковалентной связи. Примерами аогут служить галогениды элементов второй и третьей групп ПСЭ (BF 3 ,AlCl 3 ,FeCl 3 ,ZnCl 2 , катионы металлов, Н + . кислоты Льюиса в гетеролитических реакциях участвуют как электрофильные реагенты.
Основаниями Льюиса могут быть атом, молекула или анион, имеющие пару валентных электронов, которые они должны предоставить партнеру для образования ковалентной связи. Основания Льюиса представляют собой нуклеофильные реагенты. К ним относятся амины, спирты, простые эфиры, тиолы, тиоэфиры, соединения, содержащие π-связи или систему сопряженных π-связей.
Важным следствием теории Льюиса является то, что любое органическое соединение можно представить как кислотно-основной комплекс. Например, этилиодид можно рассматривать как комплекс, состоящий из этил-катиона С 2 Н 5 + (кислота Льюиса) и иодид иона –I (основание Льюиса). Отсюда большинство реакций с этилиодидом можно классифицировать как обмен иодид-иона на другие основания Льюиса (ОН - , СN - ,NH 2 - ,H 3 C-O -) или как обмен этил-катиона на другие кислоты Льюиса (К +, Н + и др.).
Вопросы для самоконтроля
Современные представления о кислотах и основаниях.
Теория Бренстеда-Лоури. Сопряженные кислоты и основания.
Кислотные свойства органических соединений с водородосодержащими функциональными группами (спирты, фенолы, тиолы, карбоновые кислоты, амины).
Основные свойства органических соединений. π-основания и n-основания. Теория Льюиса.
Факторы, влияющие на реакционную способность кислот и оснований.
Объясните, какое из соединений проявляет более сильные кислотные свойства и почему?
а) CH 3 -SH, б) CH 3 -NH 2 , в) CH 3 -OH.
Какая кислота является более сильной и почему?
а) СH 3 COOH, б) Сl-CH 2 COOH, в) Cl 2 - CH – COOH.
Объясните, почему анилин проявляет более слабые основные свойства, по сравнению с этиламином.
9. Напишите какая из реакций доказывает более сильные кислотные свойства фенола по сравнению со спиртами:
10. В качестве антидота при отравлении солями мышьяка был предложен 2,3-димеркаптопропанол. Объясните, чем определяется его большая кислотность в сравнении с пропантриолом.
Основная
1.Н. А. Тюкавкина, Органическая химия, 2002 г. С. 100-101, 108-109.





")